
Lipfendra Brings PCSK9i Efficacy with Oral Convenience
Last week, Merck’s PCSK9 inhibitor, Lipfendra (enlicitide), was approved (1) for lowering LDL-C as an adjunct to diet and exercise in adults with hypercholesterolemia. PCSK9 inhibitors are powerful drugs that offer an add-on or monotherapy option for patients who have not reached their LDL-C goals on statins alone or who cannot tolerate statins.
Trials of other drugs, including Repatha (evolocumab, Amgen), have demonstrated that lowering LDL-C with statins or PCSK9 inhibitors can lower risk of major adverse cardiovascular events (MACE) in adults at increased risk for MACE (primary prevention) or who have already had a MACE (secondary prevention).
Lipfendra’s effect on cardiovascular outcomes is still being evaluated in the CORALreef Outcomes trial (2). Using existing data and reasonable assumptions about what future outcomes data may look like, the Equinox model can estimate how Lipfendra may measure up in primary and secondary prevention.
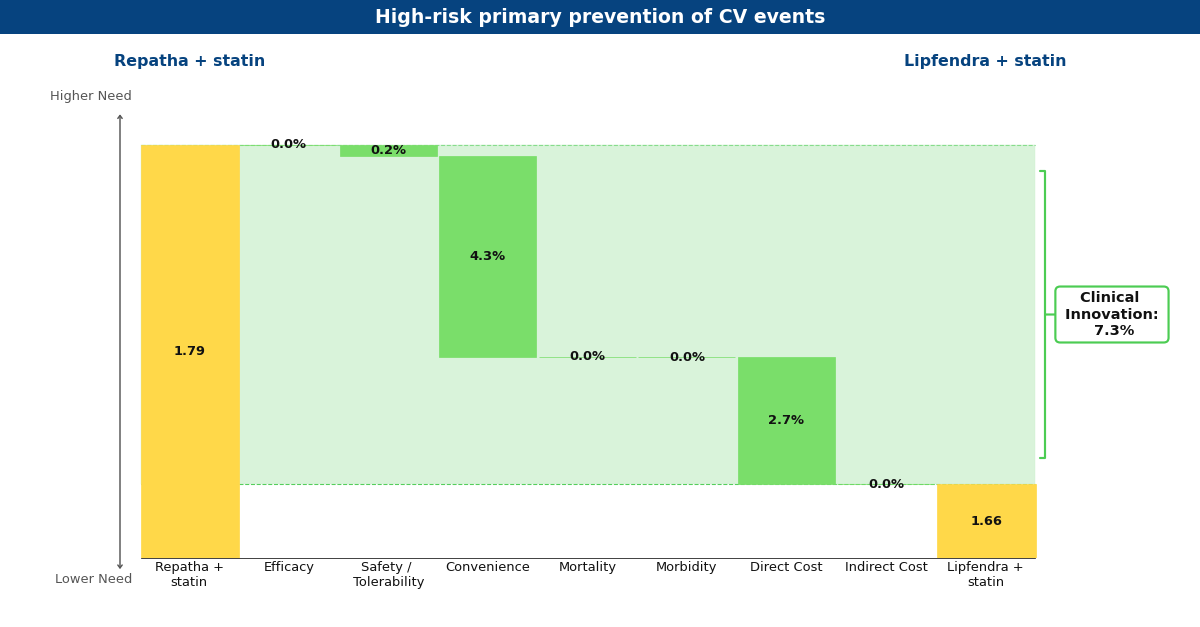
Drivers chart depicting what Equinox believes Lipfendra may look like in the future compared to Repatha for primary prevention of MACE in high-risk patients.
Lipfendra’s main driver of improvement is convenience, as it is the first approved oral PCSK9 inhibitor in a market of injectables. In Repatha’s Phase 3 MACE prevention trials (3, 4) and Lipfendra’s Phase 3 hypercholesterolemia trial (5), these drugs appeared to have a similar net-of-placebo effect on LDL-C reduction and both were well-tolerated. Based on these data, Equinox believes if Lipfendra were to be approved for primary and secondary prevention, it would be based on data demonstrating a similar tolerability and relative risk reduction in MACE as seen with Repatha.
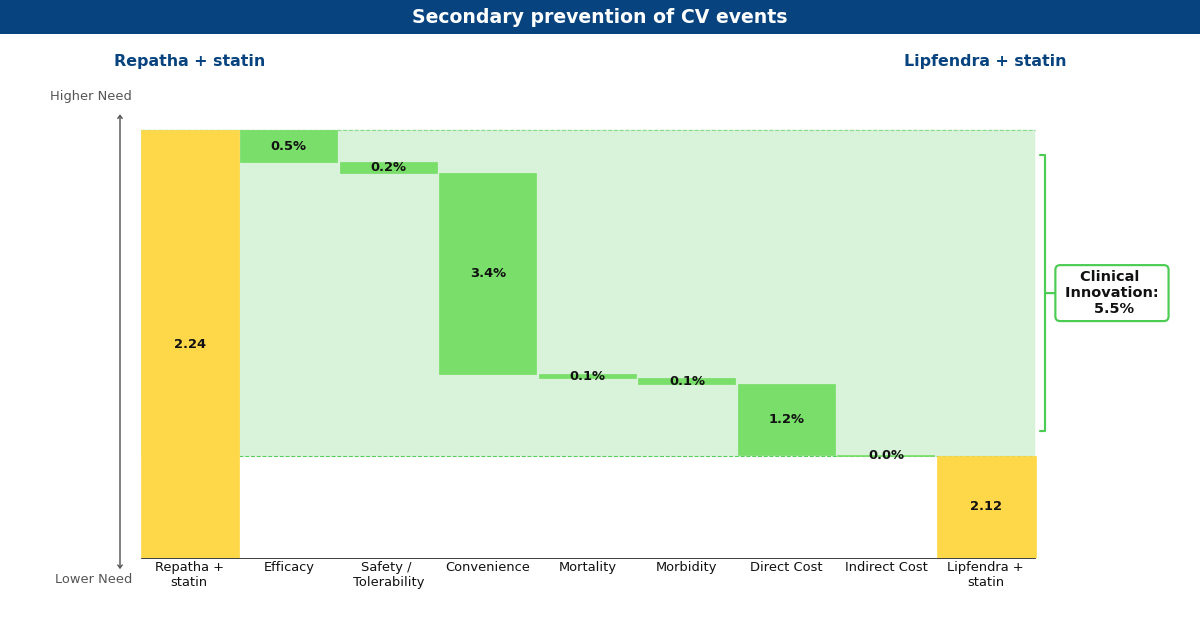
Drivers chart depicting what Equinox believes Lipfendra may look like in the future compared to Repatha for secondary prevention of MACE
Currently, Lipfendra has a substantially lower annual WAC than Repatha, resulting in overall innovation scores of 7.3% and 5.5% in primary and secondary prevention, respectively. We expect that the arrival of biosimilar Repatha will degrade this advantage somewhat, but Lipfendra would remain differentiated and commercially successful in both populations (at 6.1% and 5.0%).
Oveporexton Could be a Game-changer for Narcolepsy Type 1
Narcolepsy is a rare neurological disorder that affects an estimated 200,000 Americans [1], and about half of those people have narcolepsy type 1 (NT1) [2]. While both NT1 and NT2 cause sleep attacks and excessive daytime sleepiness, NT1 also causes cataplexy: episodes of sudden muscle weakness often triggered by intense emotions. Not only does NT1 severely impact quality of life, but it can also be dangerous when cataplexy episodes or sleep attacks occur during activities such as driving or operating machinery [3].
In February 2026, the FDA accepted the NDA for oveporexton, Takeda’s investigational agent, and granted it Priority Review. Instead of simply managing symptoms, oveporexton is a potentially first-in-class OX2R-selective agonist that targets the orexin deficiency that causes NT1 [4].
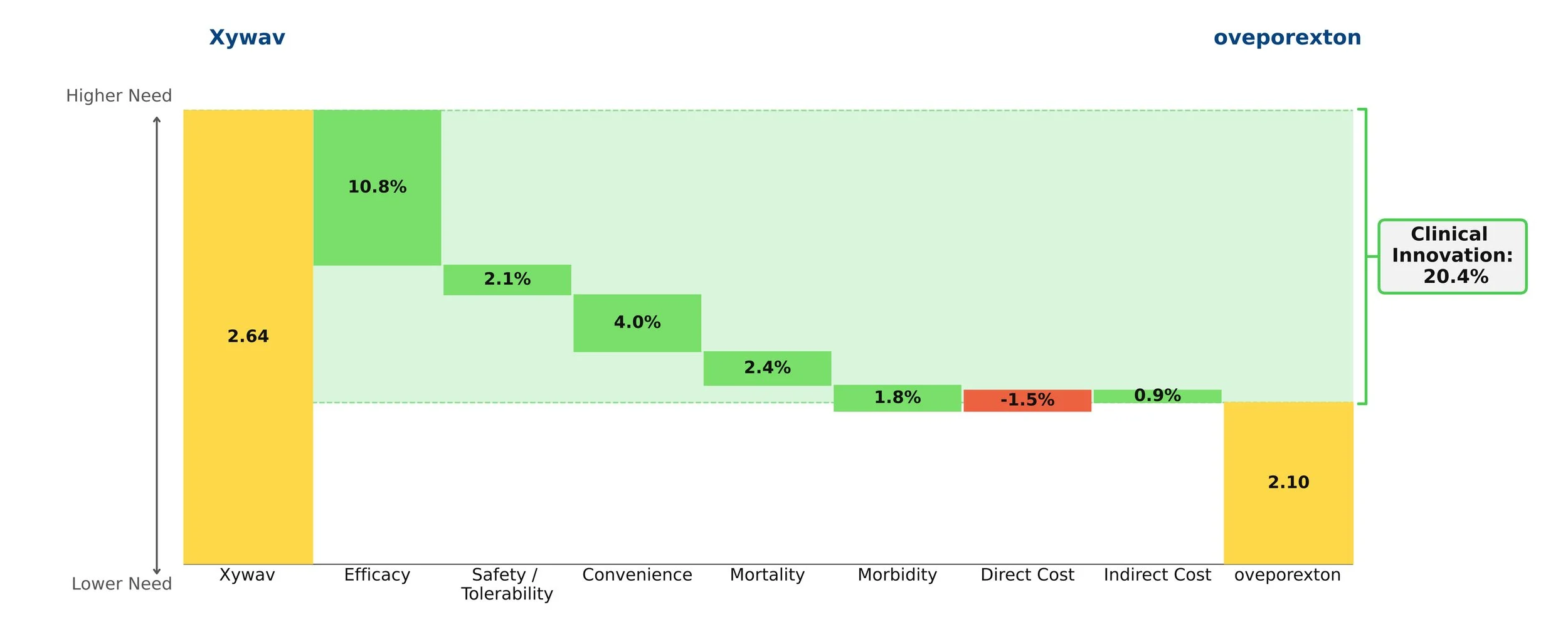
Oveporexton yields an exceptional clinical innovation score of 20.4% over standard-of-care Xywav when it is priced at a modest premium. Based on currently available data, we view oveporexton as a more efficacious and convenient option, and, if launched at this price point, expect it will take a commanding lead of the NT1 market.
Gene Therapies for Sickle Cell Disease: Expensive but Worth It
Sickle cell disease affects roughly 100,000 Americans and is more common among African American and non-Hispanic Black people [1]. About 20,000 suffer from recurrent vaso-occlusive crises (VOCs), making them ideal candidates for the novel gene therapies Casgevy (exagamglogene autotemcel, Vertex) and Lyfgenia (lovotibeglogene autotemcel, bluebird bio, rebranded as Genetix Biotherapeutics) [2]. Priced at $2.2 million and $3.1 million, respectively, these drugs are highly innovative and – we conclude – worth the high price tags if payers can figure out how to foot the bill.
Approved in December 2023, Casgevy is a CRISPR/CAS9-based therapy, the first of its kind. Its administration procedure is similar to that of a stem cell transplant and it is incredibly efficacious, with 93% of patients in the clinical trial remaining VOC-free 12 months after treatment [3]. Casgevy also boasts an approval in transfusion-dependent beta-thalassemia, a rare blood disorder that affects only 1,300-1,500 people in the US [4]. Lyfgenia is similarly efficacious and works via a lentiviral vector to insert a functional copy of the beta globin gene to increase the production of normal hemoglobin.
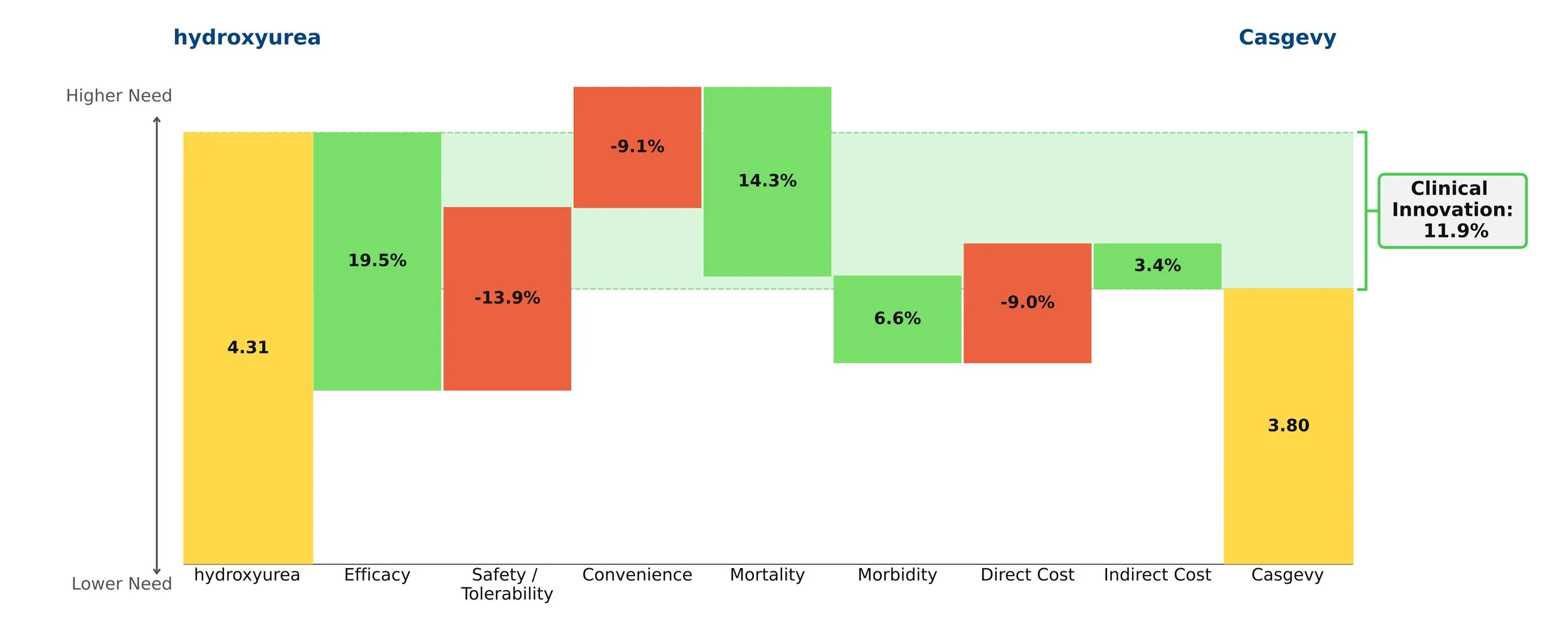
Both therapies demonstrate high clinical innovation when compared to the standard of care, hydroxyrurea, with a direct cost amortized over three years. Amortization is based off clinical data demonstrating that patients who achieve VOC-free status over 12 months remain VOC-free for approximately 3 years [5, 6]. This assessment may change as more long-term data becomes available. With the life expectancy of sickle cell disease patients being far shorter than for those without the disease, these therapies have the potential to substantially close that gap, with a University of Washington study finding a benefit of approximately 17 years of increased life expectancy from the gene therapies [7]. Our model captures a dramatic 85% reduction in mortality to align with this. These therapies are even more impressive when considering the high unmet need of sickle cell disease, in addition to the societal and indirect cost savings they may bring.
However, the issue of paying for these high-priced therapies looms large. An analysis conducted by the Institute for Clinical and Economic Review (ICER) in 2023 concluded that they are cost-effective at a price range of $1.5-$2 million [8]. At $2.2 million, Casgevy is pushing that limit, and at $3.1 million, Lyfgenia is well out of the range. With these steep price tags, it will be a challenge for payers to figure out how to pay for them, especially considering that a large percentage of the patient population is underserved and on Medicaid [9]. Currently, CMS has proposed an outcomes-based pricing scheme (CGT access model) that individual states can opt into. Only patients enrolled in Medicaid could benefit from the model, which began in early 2025.
This model has the potential to reduce the cost for states to bear, as CMS is the central negotiator for all states and will be providing federal funding for the treatment. States can choose which gene therapies to cover [10]. Based on our analysis, we believe that covering Casgevy is more reasonable than Lyfgenia, but having more options could be beneficial for patients, even with Lyfgenia’s black box warning for hematologic malignancy that demands long-term monitoring indefinitely [11]. Manufacturers will be encouraged to provide rebates and reimburse accordingly in cases where clinical performance falls short. The initiative will also be collecting data over eleven years, with an outcomes-based agreement term of one to six performance years, which will provide further insight into navigating these expensive gene therapies [12]. The model does not include private insurance plans for those not enrolled in Medicaid. Patients on private insurance plans may face additional requirements for treatment, such as meeting a specific threshold of number of VOCs per year, and a baseline level of decent health.
Gene therapies have limits; they are not foolproof cures. Not all cells can uptake the edits, there may be off-target gene editing effects, they are not effective for every patient, and immune system responses may limit efficacy and compromise health [13]. The treatment journey is also time-consuming, with the Casgevy website stating that it can take up to one year [14]. Since long-term data are not currently available, we must learn as we go, but it is clear that Casgevy and Lyfgenia are an important milestone in the cell and gene therapy space.
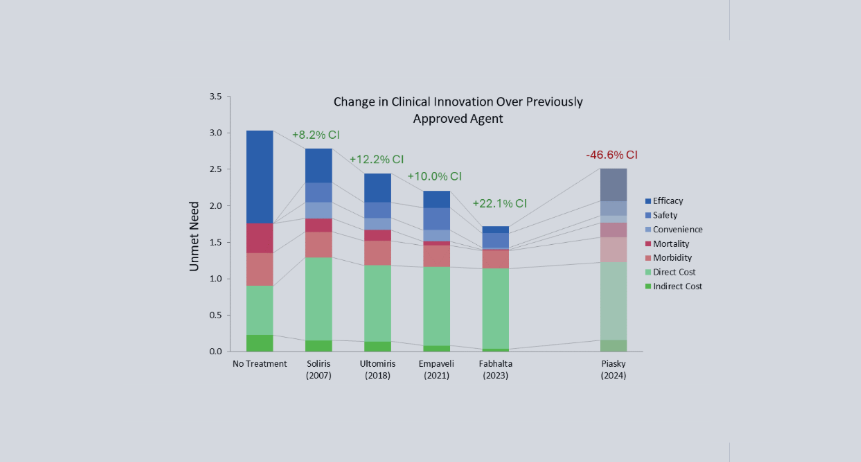
Using Equinox Drivers Charts to Understand Fabhalta's Success in PNH
Paroxysmal Nocturnal Hemoglobinuria (PNH) is rare blood disorder estimated to have a global prevalence of 12 to 13 per 1,000,000 people [1]. This condition can potentially lead to kidney disease, hemolytic anemia, and even life-threatening blood clots [2]. Until the C5-inhibitor Soliris (Alexion, eculizumab) was approved in 2007, there were no approved therapies for PNH [3].
While Soliris, along with Alexion’s other C5 inhibitor, Ultomiris (ravulizumab, which was approved for PNH in 2018 [4]), are still considered standards of care, the market has grown with the approvals of Empaveli (Apellis, pegcetacoplan), Fabhalta (Novartis, iptacopan), and PiaSky (Roche, crovalimab). Using Equinox drivers charts can help us understand how these newer entries compare to Alexion’s drugs.
Equinox characterizes a drug’s improvement relative to a previous standard of care or another drug with a Clinical Innovation (CI) score, which quantifies how much the new agent lowers unmet need in the indication. For frame of reference, products with >2% CI score are typically commercially viable, products with >5% CI score are commercially successful (top 3-4 in market), and products with >10% CI score are market dominators.
Fabhalta has a whopping CI score of 29.9% when compared to Ultomiris. Though Fabhalta being the first oral agent in this indication is an improvement on its own, its substantial efficacy benefits flow through to improve morbidity and mortality across the board. Although Fabhalta has the highest WAC ((Wholesale Acquisition Cost) among the PNH drugs in this analysis, this is offset by reductions in hospitalization and associated medical costs.
While Empaveli improves upon Ultomiris’s efficacy, it does not do so to the same extent Fabhalta does. Additionally, while Empaveli can be administered at home, the frequency of administration is increased, offsetting some of its convenience benefit. In most cases, a 10.0% CI score would be expected to be a market dominator, but because it still falls far short of Fabhalta, this would be an exception.

Finally, although Piasky is more convenient than Ultomiris, this is not enough to overcome its marginally reduced efficacy at the current WAC. We do not expect it to be competitive in this market, especially against newer drugs that reduce unmet need further, such as Fabhalta and Empaveli.
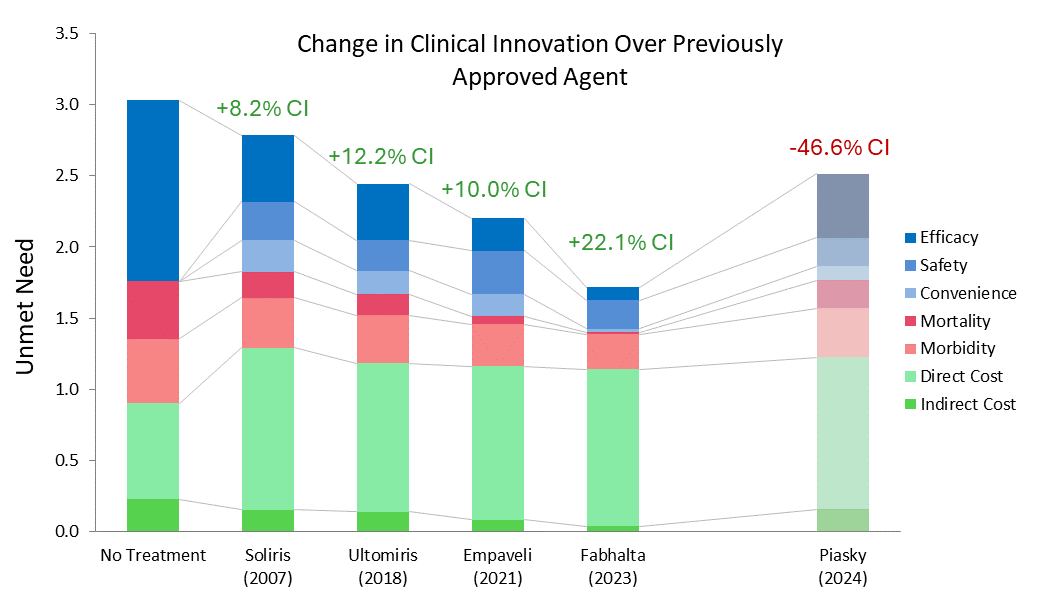
Soliris 2007 reflects Soliris WAC at launch; all costs inflated to 2025 USD.
Equinox also acknowledges the approval of Voydeya in this indication, though it was not included in this analysis due to it being approved as an add-on therapy for Soliris or Ultomiris, not a monotherapy.
Journavx, a vast improvement over opioids in moderate acute pain
Journavx’s clinical data in its first approved indication suggest it will be a game-changer in moderate acute pain management as it reduced medical need by more than 15% compared to hydrocodone + acetaminophen. (Figure 1) For reference, new drugs with 10%+ innovation over the standard of care generally achieve significant patient share. (Figure 2)
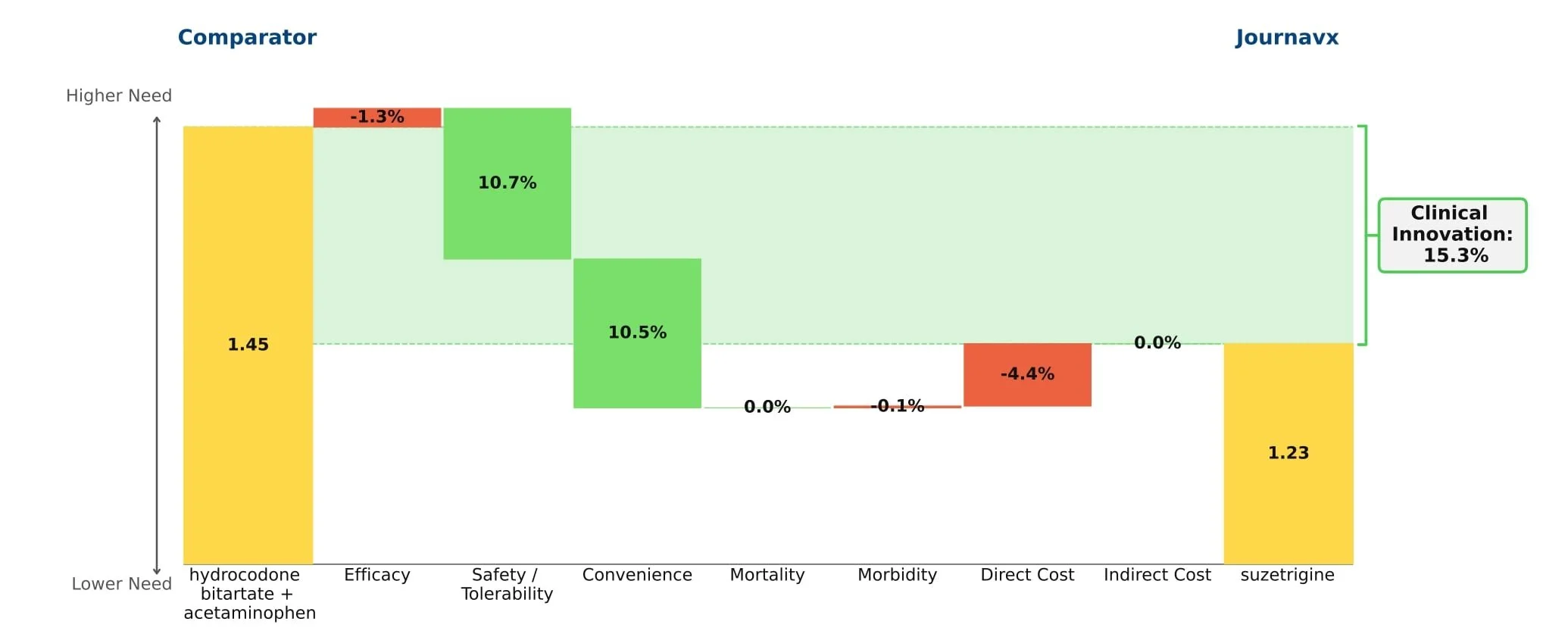
Figure 1: Drivers of Journavx's Clinical Innovation
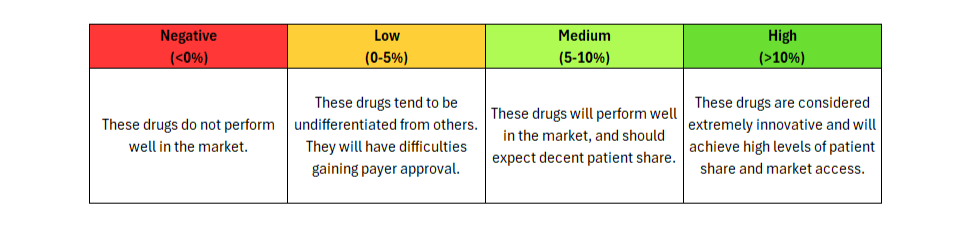
Figure 2: Understanding Clinical Innovation
Journavx (suzetrigine), a sodium channel blocker from Vertex Pharmaceuticals, was approved on January 30, 2025 in moderate to severe acute pain, based on a pair of clinical trials that compared suzetrigine to a low dose opioid, 5 mg hydrocodone bitartrate in combination with 325 mg acetaminophen (HB/APAP), in patients that underwent abdominoplasty or bunionectomy. In these trials, suzetrigine offered reduced side effects while of course lacking the black box warning and schedule II drug status of hydrocodone. [1] These improvements, along with a slight boost from less frequent dosing, result in a substantial benefit in safety and convenience over HB/APAP and account for all of Journavx’s clinical innovation in Equinox Group’s analysis.
In addition to these strong advantages, suzetrigine held its own in terms of efficacy, with an improvement over placebo in the time-weighted reduction of pain intensity similar to that of HB/APAP (10% improvement vs. 11% in the HB/APAP group) in a pooled analysis of the two studies. The higher cost of suzetrigine claws back some of the benefits, but the net impact is a clinical innovation score of 15.3%, which is characteristic of a market-dominator. [2, 3]
Given the ongoing opioid epidemic in America, addiction free pain relief remains a significant unmet need. In 2024 alone, it was estimated that 4.8 million Americans had an opioid use disorder in the past year and 7.6 million had misused prescription opioids over that same period. [4] While the current clinical importance of these medications is undeniable, it has long been agreed upon that the development of an alternative to opioids, especially for populations only experiencing moderate pain, is essential.
While Journavx proves significantly advantageous in the management of acute pain following minor surgeries, it remains to be seen how it stacks up against opioids in populations with more severe pain. In addition to exploring this further, Vertex is currently conducting a trial in diabetic peripheral neuropathy, suggesting we may see a future label expansion into the chronic pain space as well. But given these impressive initial results, Equinox Group suspects that suzetrigine will carve out a sizable share of the overall pain market.
[1] JOURNAVX Prescribing Information. FDA. Accessed July 11, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219209Orig1s000lbl.pdf
[2] Vertex Pharmaceuticals Incorporated, Evaluation of Efficacy and Safety of Suzetrigine for Acute Pain After an Abdominoplasty. ClinicalTrials.gov identifier: NCT05558410. Last updated: July 1, 2025. Accessed August 29, 2025. https://clinicaltrials.gov/study/NCT05558410
[3] Vertex Pharmaceuticals Incorporated, Evaluation of Efficacy and Safety of VX-548 for Acute Pain After a Bunionectomy. ClinicalTrials.gov identifier: NCT05553366. Last updated: December 16, 2024. Accessed July 11, 2025. https://clinicaltrials.gov/study/NCT05553366
[4] Substance Abuse and Mental Health Services Administration. (2025). Key substance use and mental health indicators in the United States: Results from the 2024 National Survey on Drug Use and Health (HHS Publication No. PEP25-07-007, NSDUH Series H-60). Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration.https://www.samhsa.gov/data/sites/default/files/reports/rpt56287/2024-nsduh-annual-national-report.pdf
Xolair for food allergies: effective therapy at a high price
Food allergies affect 5-10% of the American population. Their prevalence continues to increase, with significant quality-of-life impairments for patients and their families[1]. It is a unique indication in that patients are not chronically ill, but acute illness can occur within minutes even with proper precautions. Death is extremely rare[2]. For decades, the space has seen little in the way of treatment, with the cornerstone of management being strict allergen avoidance. Oral immunotherapy is an option to desensitize patients to their allergens, but it is burdensome and carries risk of allergic reactions and high discontinuation rates[3]. Xolair (omalizumab, anti-IgE antibody, Genentech/Novartis) offers high clinical benefit, safely increasing the amount of allergen that patients can tolerate without experiencing symptoms of severe allergic reactions that can occur through accidental exposure.

Xolair is incredibly efficacious in this indication, with modest safety/tolerability and convenience drawbacks. Even though it comes with a black box warning for anaphylaxis, safety is a minor concern in practice- it boasts a mild side effect profile and its safety is well established, as it has been on the market for more than 20 years in other indications.
Xolair also confers benefit in common comorbid allergic conditions, such as rhinitis, asthma, and eczema. Xolair comes as a metered-dose autoinjector or prefilled syringe that can be administered at home approximately every other week or every four weeks, depending on baseline IgE levels and body weight, and must be administered indefinitely for continued benefit. Typically, drugs with clinical innovation of 5% or more go on to achieve reasonably strong patient share. Xolair in this indication has very high clinical benefits, but at a steep price of around $100,000 a year. We anticipate strong interest from patients and physicians, but strong push-back from payers.
Xolair will likely become a candidate for biosimilar development when it goes off patent within the next few years, which will likely mitigate the high price and expand access in this poorly served indication.
[1] https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2022/20220126.htm
Monoclonal antibodies for Alzheimer’s disease: questionable clinical benefit at a high price
With novel treatments for Alzheimer’s disease dominating the headlines, the most recent being donanemab with the FDA requesting an independent advisory committee to review its safety and efficacy[1], questions remain about their clinical benefit. As the number of patients with Alzheimer’s disease in the US is projected to grow in the coming years, so does the need for a paradigm-changing treatment. However, our model finds that the new monoclonal antibodies offer little clinical innovation over the old standard of care, donepezil. Leaving aside price, their disadvantages in safety/tolerability and dosing outweigh their modest efficacy gains.
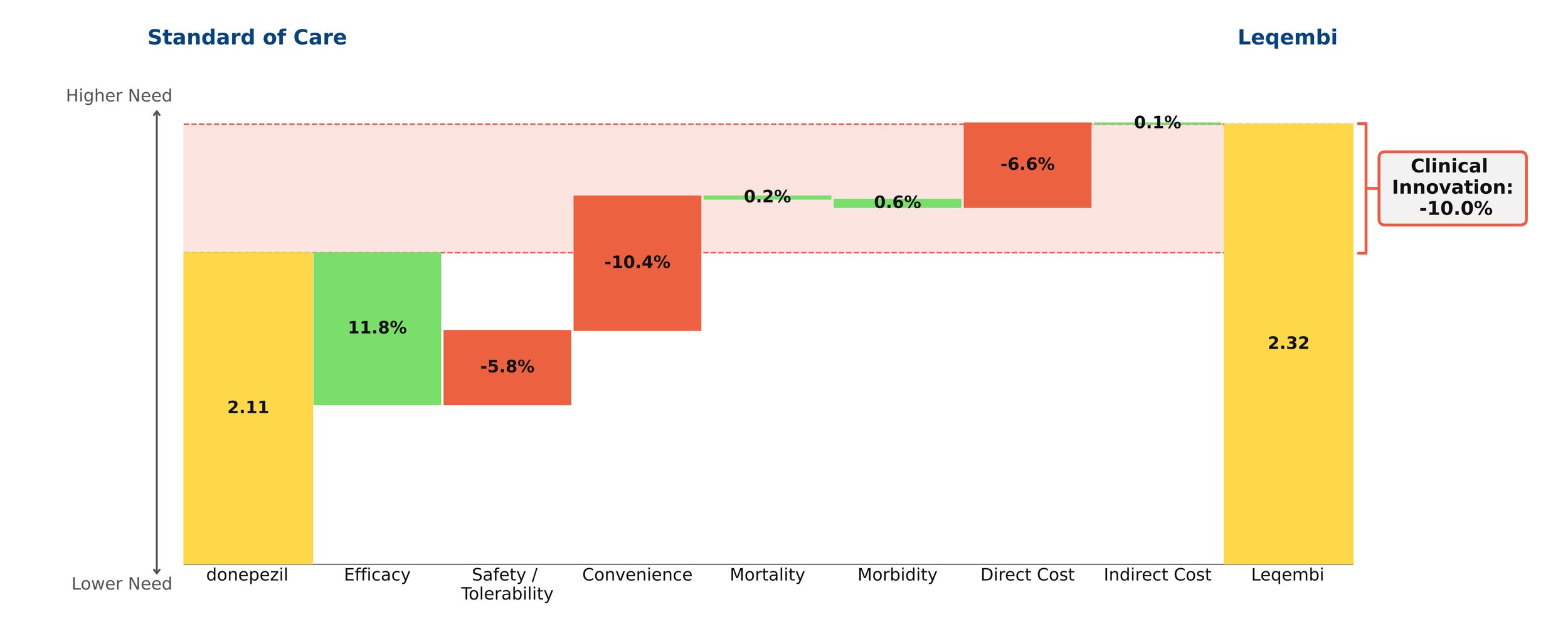
Despite remaining on the market, unlike its ill-fated predecessor Aduhelm, Leqembi shows negative clinical innovation over donepezil of -10.0% in our framework. This is little better than Aduhelm, which shows -11.0% clinical innovation vs. donepezil. As shown above, much of the negative clinical innovation is driven by convenience. Leqembi is given as a one-hour IV infusion every two weeks, compared with donepezil’s once-a-day pill. While Leqembi offers an efficacy benefit measured by standard measures of cognitive decline in AD, this pales in comparison to the inconvenience of the regimen, combined with high cost, high prevalence of potentially serious side effects and a black box warning for amyloid-related imaging abnormalities (ARIA). Questions also remain about how clinically meaningful the improved efficacy is: it is unclear how much patients and their families can appreciate a slowing in cognitive decline of about 6 months for the fraction of patients that experience it.[2]
Even though little separates Leqembi from Aduhelm in our framework, Leqembi has managed to achieve modest sales – around $10.1 million in 2023, still well below the original Eisai projections of $28 million for the year.[3] This is despite the fact that the Institute for Clinical and Economic Review (ICER) deemed its current annual WAC price of around $26,000 to offer low long-term value for money, suggesting a more appropriate price is around $10,000 annually.[4]
In spite of these equivocal results, in-class Eli Lilly drug donanemab was originally expected to be approved this year, with some analysts projecting blockbuster sales of more than $1 billion by 2025.[5] However, our model suggests donanemab will offer slightly lower clinical innovation than Leqembi or Aduhelm, owing to a poorer safety/adverse event profile and higher rates of ARIA.
Leqembi is currently trialing in subcutaneous form, as is donanemab. More convenient dosing would improve the outlook for these agents, but they will still face the headwinds of modest efficacy and significant safety concerns, as well as burdensome monitoring/imaging requirements. With all these considerations, it is difficult to believe that Eisai will hit its revenue target of $8.8 billion by 2032.[6]
[1] Edited to note 3/8/24 New York Times article
[2] https://memory.ucsf.edu/lecanemab
[3] https://www.biopharmadive.com/news/eisai-leqembi-alzheimers-target-revenue-earnings/706668/, https://www.pharmalive.com/eisai-sees-dramatic-increase-in-leqembi-uptake-following-full-fda-approval/, https://www.reuters.com/business/healthcare-pharmaceuticals/eisai-expects-alzheimers-drug-rake-revenue-665-mln-by-march-2023-11-07/
Evrysdi: A Potential Game-Changer for SMA Type 1 Patients
Spinraza was the first effective treatment option for Spinal Muscular Atrophy (SMA) Type 1. Zolgensma and Evrysdi are reshaping the market because of their superior efficacy.
SMA Type 1 is a rare, debilitating genetic disorder, affecting about 1 in 10,000 live births, with symptoms beginning before six months of age. Historically, infants born with this disorder could not sit without support. Most were expected to live two years or less.
The FDA approval of Spinraza (nusinersen, Biogen) in 2016 finally offered hope to families. The ENDEAR trial of Spinraza had such positive results that it was terminated early to give all participants access to Spinraza in an open-label extension (SHINE). Since then, Zolgensma (onasemnogene abeparvovec, Novartis, approved 2019) and Evrysdi (risdiplam, Roche, approved 2020) have emerged as alternative treatment options.
Like Spinraza, Evrysdi is a chronic treatment. However, it offers several benefits over Spinraza. In infantile-onset, symptomatic SMA Type 1 patients less than two years old, Evrysdi has noticeably higher efficacy (event-free survival and motor milestone response). These gains reduce mortality and morbidity. Evrysdi is a daily oral therapy, more convenient than Spinraza’s intrathecal bolus route.
Evrysdi is dosed by kg/bodyweight, with an annual price cap not to exceed that of treating a 44 lb child, complicating any simple cost comparison. However, even at its maximum price, the annual cost of Evrysdi is less than that of maintenance Spinraza.

In the Equinox model, Evrysdi offers 15.1% clinical innovation versus Spinraza. Historically, drugs that have scored >10% clinical innovation have become market dominators, and so far, annual sales of Evrysdi have trended in this direction.

One way that Spinraza has an edge over Evrysdi is its quantity of supporting data, since Evrysdi has not been around as long to establish a track record.
Zolgensma is another promising treatment option for SMA type 1. Unlike Spinraza and Evrysdi, Zolgensma is a single-dose gene therapy. At approximately $2.25 million 2023 USD (WAC, Micromedex), its price tag has been criticized in the past. However, Novartis offers different payment plans, including outcome-based and over-time payment plans, both up to 5 years.

Spinraza and Evrysdi (at maximum dose) overtake that $2.25 million mark after about 5 and 6 years of treatment, respectively. However, this calculation ignores other direct costs (such as hospitalization) and the potential for additional therapies if the initial treatment is insufficient. This added cost is worth considering since recent data shows that 7.5 years after dosing, almost one-third of Zolgensma-treated patients had received follow-up treatment. Unlike Spinraza and Evrysdi, a patient cannot simply switch off of Zolgensma if it does not perform as expected. Discounts, negotiations, and payer willingness to sponsor an expensive one-time therapy like Zolgensma also complicate the picture.
In summary, in a head-to-head comparison against Spinraza, Evrysdi is the clear winner. This sets it up to become a future standard-of-care as data continue to accumulate. Zolgensma is also innovative over Spinraza. However, it will likely continue to face challenges as payers continue to negotiate the best way to finance an expensive one-time treatment where a cost-effective outcome cannot always be guaranteed.
Prospects for ulotaront in schizophrenia
Conclusion: Though ulotaront offers an improved safety/tolerability profile over the generic antipsychotic risperidone for patients with schizophrenia, its likely price would consign it to later-line use if approved.
Antipsychotics such as risperidone, olanzapine, and quetiapine provide efficacy for patients with schizophrenia, but come with substantial safety/tolerability baggage. However, Ulotaront (Sunovion), an antipsychotic in phase 3 clinical trials, may reduce unmet need by providing efficacy comparable to current therapies with fewer tolerability issues.
We modeled risperidone as the standard of care, as it gave the lowest unmet need score in our framework. Efficacy values on the Positive and Negative Syndrome Scale (PANSS) in our analysis are from short-term trials, but efficacy may differ in long-term settings and uncontrolled patient populations.

While ulotaront did not reduce PANSS scores as much as risperidone did, it had a lower discontinuation rate, giving it a slight edge over risperidone in efficacy. Ulotaront appears to provide acceptable efficacy with improved tolerability over the commonly used atypical antipsychotics.
However, we expect ulotaront’s cost will limit its share. We projected its annual wholesale acquisition cost (WAC) at about $17,000, equal to that of Lybalvi (olanzapine/samidorphan, Alkermes), a schizophrenia drug with a similar therapeutic profile that entered the US market in late 2021. Generic risperidone’s yearly cost is about $130. The assumed price diminishes ulotaront’s clinical innovation by -6.9%. The result is overall minimal clinical innovation of 1.8%. History shows that new drugs need to hit 5% clinical innovation to have good commercial prospects.

If we compare ulotaront with Lybalvi, ulotaront has 2.7% clinical innovation over Lybalvi, based on a slightly better side effect profile and marginally better efficacy.
We predict that ulotaront will occupy a niche similar to Lybalvi’s. Both drugs have efficacy like the second-generation antipsychotics, a high price penalty, and notably improved safety and tolerability. Generics will remain the top first-line choice owing to price. However, even with low clinical innovation compared to risperidone, we expect ulotaront and Lybalvi will be important for patients who fail or cannot tolerate generic antipsychotics.
Lybalvi’s team is already thinking about competing against generics, expecting that the frequent drug-switching (usually driven by adverse events) seen in schizophrenia will get their drug tested in many patients. Sunovion will likely adopt a similar strategy with ulotaront.
Trikafta: A Major Step Forward for Cystic Fibrosis Patients
Cystic fibrosis (CF) affects more than 30,000 patients in the US. Recent treatments for CF focus on modulating the CFTR gene, with Vertex Pharmaceuticals’ Kalydeco (ivacaftor), Orkambi (lumacaftor/ivacaftor), and Symdeko (tezacaftor/ivacaftor) as the only approved therapies that provide more than symptomatic relief. Those agents, however, either are indicated for small segments of the CF population or offer modest clinical improvements as measured in Equinox Group’s model.
Enter Vertex’s newest treatment, Trikafta (elexacaftor/tezacaftor/ivacaftor), approved in October 2019, which expands the number of CF patients who can benefit from CFTR modulators and offers significant improvement in clinical outcomes. Trikafta has a broad label for all CF patients aged 12 years and older with at least one F508del mutation (approximately 85% of CF patients in the US carry a copy of the F508del mutation). Trikafta’s approval allows Vertex to treat both the underserved heterozygous F508del population and the homozygous F508del population, where both Orkambi and Symdeko are options.
Trikafta offers very high clinical innovation in patients with the heterozygous F508del mutation, delivering a 15% improvement over Pulmozyme (dornase alfa, Genentech) — with clear gains in efficacy, mortality, and morbidity:

For these heterozygous patients, Equinox’s Rare Disease Normative Price Calculator finds Trikafta to be reasonably priced at annual US WAC of $311,500, given its level of clinical benefit.

Equinox Group’s research and predictive model for pricing agents targeted to rare diseases has found that three factors drive pricing potential:
Size of the patient population
Level of disease seriousness (mortality and morbidity), and
Clinical improvement as measured in the Equinox unmet need model
Trikafta’s published data in the homozygous population is not sufficient to allow Equinox to accurately characterize the clinical benefit for those patients, so we cannot comment on the appropriateness of its price in that population.
In Q1 of 2020, Trikafta achieved $900 million in sales. That substantial and rapid commercial success is attributable both to a much larger target population than Vertex’s older CF treatments and to its very high clinical innovation in the heterozygous F508del group.