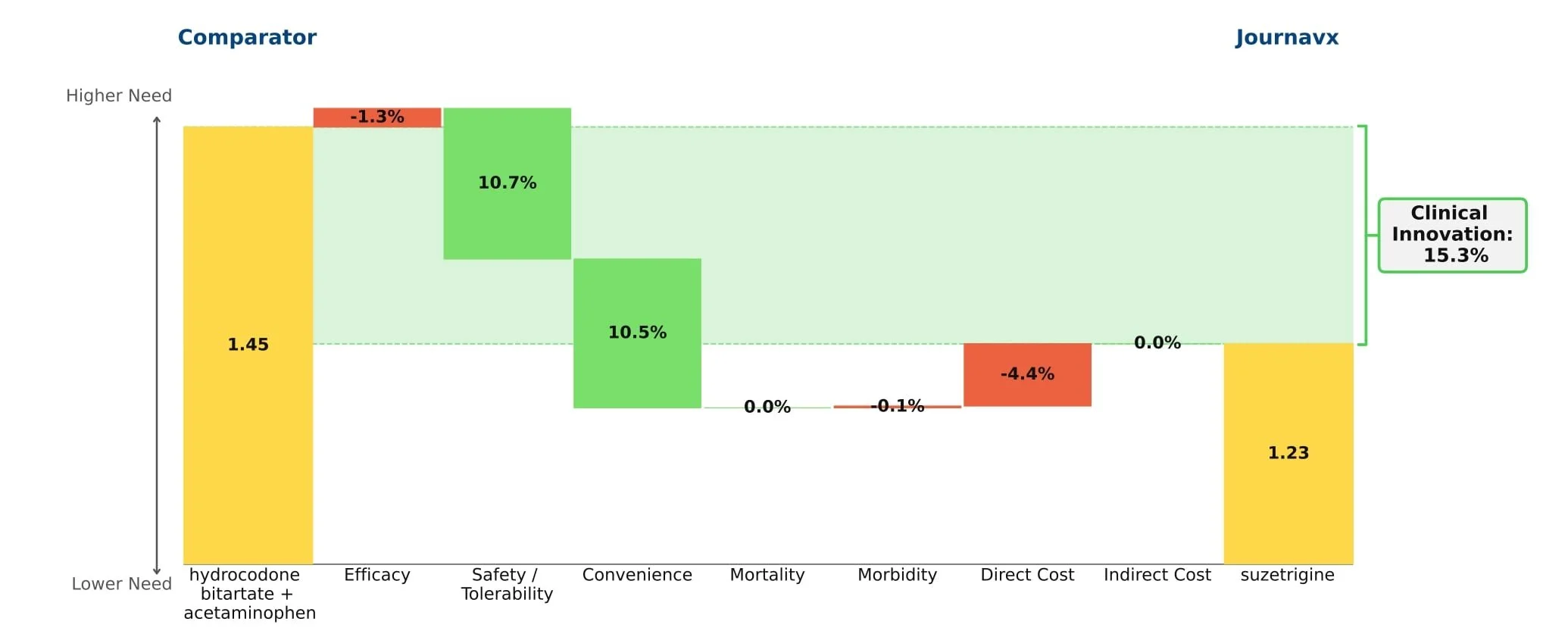
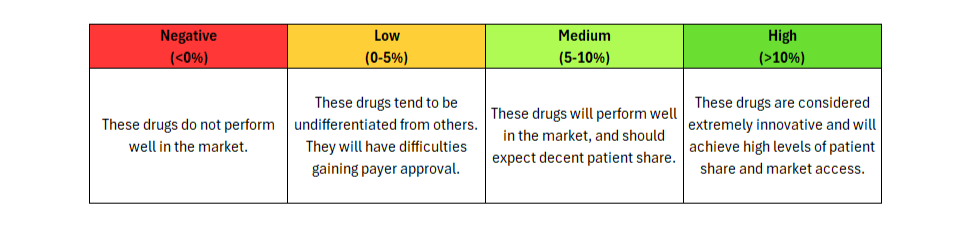
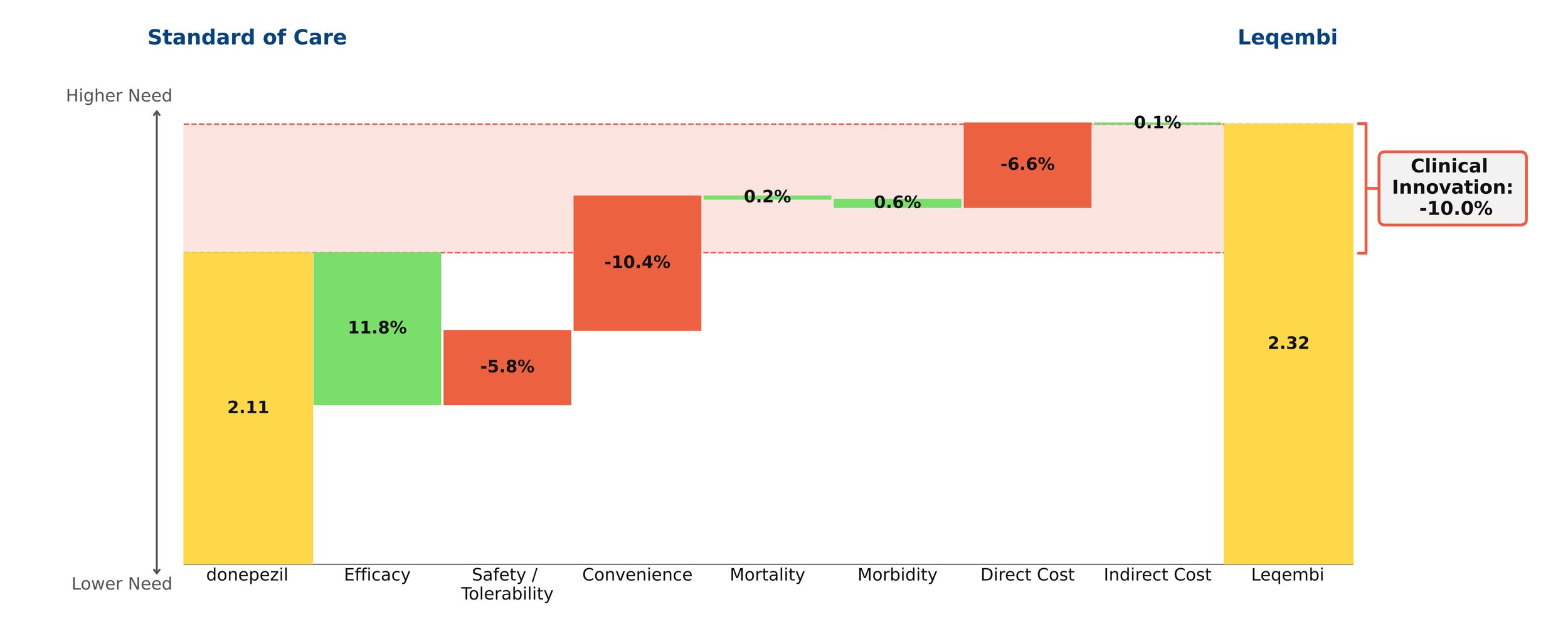
Journavx (suzetrigine), a sodium channel blocker from Vertex Pharmaceuticals, was approved on January 30, 2025 in moderate to severe acute pain, based on a pair of clinical trials that compared suzetrigine to a low dose opioid, 5 mg hydrocodone bitartrate in combination with 325 mg acetaminophen (HB/APAP), in patients that underwent abdominoplasty or bunionectomy. In these trials, suzetrigine offered reduced side effects while of course lacking the black box warning and schedule II drug status of hydrocodone. [1] These improvements, along with a slight boost from less frequent dosing, result in a substantial benefit in safety and convenience over HB/APAP and account for all of Journavx’s clinical innovation in Equinox Group’s analysis.
In addition to these strong advantages, suzetrigine held its own in terms of efficacy, with an improvement over placebo in the time-weighted reduction of pain intensity similar to that of HB/APAP (10% improvement vs. 11% in the HB/APAP group) in a pooled analysis of the two studies. The higher cost of suzetrigine claws back some of the benefits, but the net impact is a clinical innovation score of 15.3%, which is characteristic of a market-dominator. [2, 3]
Given the ongoing opioid epidemic in America, addiction free pain relief remains a significant unmet need. In 2024 alone, it was estimated that 4.8 million Americans had an opioid use disorder in the past year and 7.6 million had misused prescription opioids over that same period. [4] While the current clinical importance of these medications is undeniable, it has long been agreed upon that the development of an alternative to opioids, especially for populations only experiencing moderate pain, is essential.
While Journavx proves significantly advantageous in the management of acute pain following minor surgeries, it remains to be seen how it stacks up against opioids in populations with more severe pain. In addition to exploring this further, Vertex is currently conducting a trial in diabetic peripheral neuropathy, suggesting we may see a future label expansion into the chronic pain space as well. But given these impressive initial results, Equinox Group suspects that suzetrigine will carve out a sizable share of the overall pain market.
[1] JOURNAVX Prescribing Information. FDA. Accessed July 11, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219209Orig1s000lbl.pdf
[2] Vertex Pharmaceuticals Incorporated, Evaluation of Efficacy and Safety of Suzetrigine for Acute Pain After an Abdominoplasty. ClinicalTrials.gov identifier: NCT05558410. Last updated: July 1, 2025. Accessed August 29, 2025. https://clinicaltrials.gov/study/NCT05558410
[3] Vertex Pharmaceuticals Incorporated, Evaluation of Efficacy and Safety of VX-548 for Acute Pain After a Bunionectomy. ClinicalTrials.gov identifier: NCT05553366. Last updated: December 16, 2024. Accessed July 11, 2025. https://clinicaltrials.gov/study/NCT05553366
[4] Substance Abuse and Mental Health Services Administration. (2025). Key substance use and mental health indicators in the United States: Results from the 2024 National Survey on Drug Use and Health (HHS Publication No. PEP25-07-007, NSDUH Series H-60). Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration.https://www.samhsa.gov/data/sites/default/files/reports/rpt56287/2024-nsduh-annual-national-report.pdf